Using CellTypist for multi-label classification
This notebook showcases the multi-label classification for scRNA-seq query data using either the built-in CellTypist models or the user-trained custom models.
Only the main steps and key parameters are introduced in this notebook. Refer to detailed Usage if you want to learn more.
About multi-label cell type classification
An ideal CellTypist model is supposed to be trained from a reference atlas with a comprehensive cell type repertoire. For the built-in models, we have collected a large number of cell types; yet, the presence of unexpected (e.g., low-quality or novel cell types) and ambiguous cell states (e.g., doublets) in the query data is beyond the prediction that CellTypist can achieve with a ‘find-a-best-match’ mode. To overcome this, CellTypist provides the option of multi-label cell type classification, which assigns 0 (i.e., unassigned), 1, or >=2 cell type labels to each query cell.
Install CellTypist
[1]:
!pip install celltypist
Collecting celltypist
Using cached celltypist-1.2.0-py3-none-any.whl (5.3 MB)
Requirement already satisfied: click>=7.1.2 in /opt/conda/lib/python3.8/site-packages (from celltypist) (7.1.2)
Requirement already satisfied: scanpy>=1.7.0 in /opt/conda/lib/python3.8/site-packages (from celltypist) (1.7.1)
Requirement already satisfied: pandas>=1.0.5 in /opt/conda/lib/python3.8/site-packages (from celltypist) (1.2.3)
Requirement already satisfied: openpyxl>=3.0.4 in /opt/conda/lib/python3.8/site-packages (from celltypist) (3.0.7)
Requirement already satisfied: requests>=2.23.0 in /opt/conda/lib/python3.8/site-packages (from celltypist) (2.25.1)
Requirement already satisfied: leidenalg>=0.8.3 in /opt/conda/lib/python3.8/site-packages (from celltypist) (0.8.3)
Requirement already satisfied: scikit-learn>=0.24.1 in /opt/conda/lib/python3.8/site-packages (from celltypist) (0.24.1)
Requirement already satisfied: numpy>=1.19.0 in /opt/conda/lib/python3.8/site-packages (from celltypist) (1.20.1)
Requirement already satisfied: et-xmlfile in /opt/conda/lib/python3.8/site-packages (from openpyxl>=3.0.4->celltypist) (1.0.1)
Requirement already satisfied: python-dateutil>=2.7.3 in /opt/conda/lib/python3.8/site-packages (from pandas>=1.0.5->celltypist) (2.8.1)
Requirement already satisfied: pytz>=2017.3 in /opt/conda/lib/python3.8/site-packages (from pandas>=1.0.5->celltypist) (2021.1)
Requirement already satisfied: six>=1.5 in /opt/conda/lib/python3.8/site-packages (from python-dateutil>=2.7.3->pandas>=1.0.5->celltypist) (1.15.0)
Requirement already satisfied: chardet<5,>=3.0.2 in /opt/conda/lib/python3.8/site-packages (from requests>=2.23.0->celltypist) (4.0.0)
Requirement already satisfied: urllib3<1.27,>=1.21.1 in /opt/conda/lib/python3.8/site-packages (from requests>=2.23.0->celltypist) (1.26.3)
Requirement already satisfied: certifi>=2017.4.17 in /opt/conda/lib/python3.8/site-packages (from requests>=2.23.0->celltypist) (2020.12.5)
Requirement already satisfied: idna<3,>=2.5 in /opt/conda/lib/python3.8/site-packages (from requests>=2.23.0->celltypist) (2.10)
Requirement already satisfied: legacy-api-wrap in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (0.0.0)
Requirement already satisfied: seaborn in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (0.11.1)
Requirement already satisfied: numba>=0.41.0 in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (0.51.2)
Requirement already satisfied: joblib in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (1.0.1)
Requirement already satisfied: packaging in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (20.9)
Requirement already satisfied: scipy>=1.4 in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (1.6.1)
Requirement already satisfied: anndata>=0.7.4 in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (0.7.5)
Requirement already satisfied: h5py>=2.10.0 in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (3.1.0)
Requirement already satisfied: umap-learn>=0.3.10 in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (0.4.6)
Requirement already satisfied: tqdm in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (4.58.0)
Requirement already satisfied: statsmodels>=0.10.0rc2 in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (0.12.2)
Requirement already satisfied: sinfo in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (0.3.1)
Requirement already satisfied: patsy in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (0.5.1)
Requirement already satisfied: networkx>=2.3 in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (2.5)
Requirement already satisfied: matplotlib>=3.1.2 in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (3.3.4)
Requirement already satisfied: tables in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (3.6.1)
Requirement already satisfied: natsort in /opt/conda/lib/python3.8/site-packages (from scanpy>=1.7.0->celltypist) (7.1.1)
Requirement already satisfied: pyparsing!=2.0.4,!=2.1.2,!=2.1.6,>=2.0.3 in /opt/conda/lib/python3.8/site-packages (from matplotlib>=3.1.2->scanpy>=1.7.0->celltypist) (2.4.7)
Requirement already satisfied: cycler>=0.10 in /opt/conda/lib/python3.8/site-packages (from matplotlib>=3.1.2->scanpy>=1.7.0->celltypist) (0.10.0)
Requirement already satisfied: kiwisolver>=1.0.1 in /opt/conda/lib/python3.8/site-packages (from matplotlib>=3.1.2->scanpy>=1.7.0->celltypist) (1.3.1)
Requirement already satisfied: pillow>=6.2.0 in /opt/conda/lib/python3.8/site-packages (from matplotlib>=3.1.2->scanpy>=1.7.0->celltypist) (8.1.2)
Requirement already satisfied: decorator>=4.3.0 in /opt/conda/lib/python3.8/site-packages (from networkx>=2.3->scanpy>=1.7.0->celltypist) (4.4.2)
Requirement already satisfied: llvmlite<0.35,>=0.34.0.dev0 in /opt/conda/lib/python3.8/site-packages (from numba>=0.41.0->scanpy>=1.7.0->celltypist) (0.34.0)
Requirement already satisfied: setuptools in /opt/conda/lib/python3.8/site-packages (from numba>=0.41.0->scanpy>=1.7.0->celltypist) (49.6.0.post20210108)
Requirement already satisfied: threadpoolctl>=2.0.0 in /opt/conda/lib/python3.8/site-packages (from scikit-learn>=0.24.1->celltypist) (2.1.0)
Requirement already satisfied: get-version>=2.0.4 in /opt/conda/lib/python3.8/site-packages (from legacy-api-wrap->scanpy>=1.7.0->celltypist) (2.1)
Requirement already satisfied: stdlib-list in /opt/conda/lib/python3.8/site-packages (from sinfo->scanpy>=1.7.0->celltypist) (0.7.0)
Requirement already satisfied: numexpr>=2.6.2 in /opt/conda/lib/python3.8/site-packages (from tables->scanpy>=1.7.0->celltypist) (2.7.3)
Installing collected packages: celltypist
Successfully installed celltypist-1.2.0
[2]:
import scanpy as sc
import pandas as pd
[3]:
import celltypist
from celltypist import models
Download a scRNA-seq dataset of 500 immune cells
[4]:
adata_500 = sc.read('celltypist_demo_folder/demo_500_cells.h5ad', backup_url = 'https://celltypist.cog.sanger.ac.uk/Notebook_demo_data/demo_500_cells.h5ad')
This dataset includes 500 cells and 18,950 genes collected from different studies, thereby showing the practical applicability of CellTypist.
[5]:
adata_500.shape
[5]:
(500, 18950)
The expression matrix (adata_500.X) is pre-processed (and required) as log1p normalised expression to 10,000 counts per cell (this matrix can be alternatively stashed in .raw.X).
[6]:
adata_500.X.expm1().sum(axis = 1)[:10]
[6]:
matrix([[10000. ],
[10000.001],
[ 9999.999],
[10000.001],
[10000.003],
[ 9999.999],
[10000. ],
[10000.002],
[10000. ],
[ 9999.999]], dtype=float32)
Some pre-assigned cell type labels are also in the data, which will be compared to the predicted labels from CellTypist later.
[7]:
adata_500.obs
[7]:
| cell_type | |
|---|---|
| cell1 | Plasma cells |
| cell2 | Plasma cells |
| cell3 | Plasma cells |
| cell4 | Plasma cells |
| cell5 | Plasma cells |
| ... | ... |
| cell496 | Macro_pDC |
| cell497 | Macro_pDC |
| cell498 | Macro_pDC |
| cell499 | Macro_pDC |
| cell500 | Macro_pDC |
500 rows × 1 columns
Among the 12 cell types in this data, 10 are shared with the CellTypist built-in models. For the remaining two, Microglia is a novel cell type not covered by CellTypist (currently our models do not involve the brain), and Macro_pDC is a cell type in silico generated by blending the expression of macrophages with plasmacytoid dendritic cells.
[8]:
sc.pl.dotplot(adata_500, ['CX3CR1', 'C1QC', 'LILRA4'], groupby = 'cell_type', swap_axes = True, standard_scale = 'var', figsize = [12, 4])

Inspect the CellTypist built-in models
Download the latest CellTypist models.
[9]:
# Enabling `force_update = True` will overwrite existing (old) models.
models.download_models(force_update = True)
📜 Retrieving model list from server https://celltypist.cog.sanger.ac.uk/models/models.json
📚 Total models in list: 12
📂 Storing models in /home/jovyan/.celltypist/data/models
💾 Downloading model [1/12]: Immune_All_Low.pkl
💾 Downloading model [2/12]: Immune_All_High.pkl
💾 Downloading model [3/12]: Adult_Mouse_Gut.pkl
💾 Downloading model [4/12]: COVID19_Immune_Landscape.pkl
💾 Downloading model [5/12]: Cells_Fetal_Lung.pkl
💾 Downloading model [6/12]: Cells_Intestinal_Tract.pkl
💾 Downloading model [7/12]: Cells_Lung_Airway.pkl
💾 Downloading model [8/12]: Developing_Mouse_Brain.pkl
💾 Downloading model [9/12]: Healthy_COVID19_PBMC.pkl
💾 Downloading model [10/12]: Human_Lung_Atlas.pkl
💾 Downloading model [11/12]: Nuclei_Lung_Airway.pkl
💾 Downloading model [12/12]: Pan_Fetal_Human.pkl
All models are stored in models.models_path.
[10]:
models.models_path
[10]:
'/home/jovyan/.celltypist/data/models'
Get an overview of the models and what they represent.
[11]:
models.models_description()
👉 Detailed model information can be found at `https://www.celltypist.org/models`
[11]:
| model | description | |
|---|---|---|
| 0 | Immune_All_Low.pkl | immune sub-populations combined from 20 tissue... |
| 1 | Immune_All_High.pkl | immune populations combined from 20 tissues of... |
| 2 | Adult_Mouse_Gut.pkl | cell types in the adult mouse gut combined fro... |
| 3 | COVID19_Immune_Landscape.pkl | immune subtypes from lung and blood of COVID-1... |
| 4 | Cells_Fetal_Lung.pkl | cell types from human embryonic and fetal lungs |
| 5 | Cells_Intestinal_Tract.pkl | intestinal cells from fetal, pediatric and adu... |
| 6 | Cells_Lung_Airway.pkl | cell populations from scRNA-seq of five locati... |
| 7 | Developing_Mouse_Brain.pkl | cell types from the embryonic mouse brain betw... |
| 8 | Healthy_COVID19_PBMC.pkl | peripheral blood mononuclear cell types from h... |
| 9 | Human_Lung_Atlas.pkl | integrated Human Lung Cell Atlas (HLCA) combin... |
| 10 | Nuclei_Lung_Airway.pkl | cell populations from snRNA-seq of five locati... |
| 11 | Pan_Fetal_Human.pkl | stromal and immune populations from the human ... |
Choose the model you want to employ, for example, the model with all tissues combined containing low-hierarchy (high-resolution) immune cell types/subtypes.
[12]:
# Indeed, the `model` argument defaults to `Immune_All_Low.pkl`.
model = models.Model.load(model = 'Immune_All_Low.pkl')
Show the model meta information.
[13]:
model
[13]:
CellTypist model with 90 cell types and 5212 features
date: 2022-04-04 23:51:15.159293
details: immune sub-populations combined from 20 tissues of 19 studies
source: https://doi.org/10.1126/science.abl5197
version: v2
cell types: B cells, CD16+ NK cells, ..., pDC precursor
features: A1BG, A2M, ..., ZYX
This model contains 90 cell states.
[14]:
model.cell_types
[14]:
array(['B cells', 'CD16+ NK cells', 'CD16- NK cells', 'CD8a/a',
'CD8a/b(entry)', 'CMP', 'Classical monocytes', 'Cycling B cells',
'Cycling DCs', 'Cycling NK cells', 'Cycling T cells',
'Cycling gamma-delta T cells', 'Cycling monocytes', 'DC',
'DC precursor', 'DC1', 'DC2', 'DC3', 'Double-negative thymocytes',
'Double-positive thymocytes', 'ELP', 'ETP', 'Early MK',
'Early erythroid', 'Early lymphoid/T lymphoid',
'Endothelial cells', 'Epithelial cells', 'Erythrocytes',
'Fibroblasts', 'Follicular B cells', 'Follicular helper T cells',
'GMP', 'Germinal center B cells', 'Granulocytes', 'HSC/MPP',
'Hofbauer cells', 'ILC', 'ILC precursor', 'ILC1', 'ILC2', 'ILC3',
'Kidney-resident macrophages', 'Kupffer cells',
'Large pre-B cells', 'Late erythroid', 'MAIT cells', 'MEMP', 'MNP',
'Macrophages', 'Mast cells', 'Megakaryocyte precursor',
'Megakaryocyte-erythroid-mast cell progenitor',
'Megakaryocytes/platelets', 'Memory B cells',
'Memory CD4+ cytotoxic T cells', 'Mid erythroid', 'Migratory DCs',
'Mono-mac', 'Monocyte precursor', 'Monocytes', 'Myelocytes',
'NK cells', 'NKT cells', 'Naive B cells',
'Neutrophil-myeloid progenitor', 'Neutrophils',
'Non-classical monocytes', 'Plasma cells', 'Pre-pro-B cells',
'Pro-B cells', 'Promyelocytes', 'Regulatory T cells',
'Small pre-B cells', 'T(agonist)', 'Tcm/Naive cytotoxic T cells',
'Tcm/Naive helper T cells', 'Tem/Effector helper T cells',
'Tem/Effector helper T cells PD1+', 'Tem/Temra cytotoxic T cells',
'Tem/Trm cytotoxic T cells', 'Transitional B cells',
'Transitional DC', 'Transitional NK', 'Treg(diff)',
'Trm cytotoxic T cells', 'Type 1 helper T cells',
'Type 17 helper T cells', 'gamma-delta T cells', 'pDC',
'pDC precursor'], dtype=object)
Note that all built-in models are continuously being updated. Thus following the same procedure below, you may produce a different result when using a older or newer model.
Single-label classification by finding the best match in the model
In this section, we show the procedure of finding the most likely cell type labels from built-in models for the query dataset.
We use the default mode (mode = 'best match') in celltypist.annotate to transfer cell type labels from the model to the query dataset. With this mode on, each query cell is predicted into the cell type with the largest score/probability among all possible cell types in the model.
[15]:
# Not run; predict cell identities using this loaded model.
#predictions = celltypist.annotate(adata_500, model = model, majority_voting = True, mode = 'best match')
# Alternatively, just specify the model name (recommended as this ensures the model is intact every time it is loaded).
predictions = celltypist.annotate(adata_500, model = 'Immune_All_Low.pkl', majority_voting = True, mode = 'best match')
🔬 Input data has 500 cells and 18950 genes
🔗 Matching reference genes in the model
🧬 4715 features used for prediction
⚖️ Scaling input data
🖋️ Predicting labels
✅ Prediction done!
👀 Can not detect a neighborhood graph, will construct one before the over-clustering
⛓️ Over-clustering input data with resolution set to 5
🗳️ Majority voting the predictions
✅ Majority voting done!
By default (majority_voting = False), CellTypist will infer the identity of each query cell independently. This leads to raw predicted cell type labels, and usually finishes within seconds or minutes depending on the size of the query data. You can also turn on the majority-voting classifier (majority_voting = True), which refines cell identities within local subclusters after an over-clustering approach at the cost of increased runtime.
The results include both predicted cell type labels (predicted_labels), over-clustering result (over_clustering), and predicted labels after majority voting in local subclusters (majority_voting). Note in the predicted_labels, each query cell gets its inferred label by choosing the most probable cell type among all possible cell types in the given model.
[16]:
predictions.predicted_labels
[16]:
| predicted_labels | over_clustering | majority_voting | |
|---|---|---|---|
| cell1 | Plasma cells | 13 | Follicular B cells |
| cell2 | Plasma cells | 6 | Plasma cells |
| cell3 | Plasma cells | 12 | Plasma cells |
| cell4 | Plasma cells | 6 | Plasma cells |
| cell5 | Plasma cells | 6 | Plasma cells |
| ... | ... | ... | ... |
| cell496 | pDC | 9 | Macrophages |
| cell497 | Macrophages | 18 | pDC |
| cell498 | Macrophages | 9 | Macrophages |
| cell499 | Macrophages | 9 | Macrophages |
| cell500 | pDC | 9 | Macrophages |
500 rows × 3 columns
Transform the prediction result into an AnnData.
[17]:
# Get an `AnnData` with predicted labels embedded into the cell metadata columns.
adata = predictions.to_adata()
Compared to adata_500, the new adata has additional prediction information in adata.obs (predicted_labels, over_clustering, majority_voting and conf_score). Of note, all these columns can be prefixed with a specific string by setting prefix in to_adata.
[18]:
adata.obs
[18]:
| cell_type | predicted_labels | over_clustering | majority_voting | conf_score | |
|---|---|---|---|---|---|
| cell1 | Plasma cells | Plasma cells | 13 | Follicular B cells | 0.996313 |
| cell2 | Plasma cells | Plasma cells | 6 | Plasma cells | 0.999478 |
| cell3 | Plasma cells | Plasma cells | 12 | Plasma cells | 0.999957 |
| cell4 | Plasma cells | Plasma cells | 6 | Plasma cells | 0.996070 |
| cell5 | Plasma cells | Plasma cells | 6 | Plasma cells | 0.998888 |
| ... | ... | ... | ... | ... | ... |
| cell496 | Macro_pDC | pDC | 9 | Macrophages | 0.187152 |
| cell497 | Macro_pDC | Macrophages | 18 | pDC | 0.849831 |
| cell498 | Macro_pDC | Macrophages | 9 | Macrophages | 0.809677 |
| cell499 | Macro_pDC | Macrophages | 9 | Macrophages | 0.937306 |
| cell500 | Macro_pDC | pDC | 9 | Macrophages | 0.612069 |
500 rows × 5 columns
adata (If a pre-calculated neighborhood graph is already present in the AnnData, this graph construction step will be skipped).[19]:
# If the UMAP or any cell embeddings are already available in the `AnnData`, skip this command.
sc.tl.umap(adata)
Visualise the prediction results.
[20]:
sc.pl.umap(adata, color = ['cell_type', 'majority_voting'], legend_loc = 'on data')

As the images show, with the default mode, Microglia is predicted as a mixture of cell types, and Macro_pDC is mostly predicted as Macrophages.
[21]:
pd.crosstab(adata.obs.cell_type, adata.obs.majority_voting).loc[['Microglia','Macro_pDC']]
[21]:
| majority_voting | DC | DC1 | Endothelial cells | Follicular B cells | Kupffer cells | Macrophages | Mast cells | Naive B cells | Neutrophil-myeloid progenitor | Plasma cells | gamma-delta T cells | pDC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cell_type | ||||||||||||
| Microglia | 2 | 14 | 1 | 0 | 0 | 31 | 0 | 6 | 1 | 0 | 0 | 5 |
| Macro_pDC | 0 | 0 | 0 | 0 | 0 | 30 | 0 | 0 | 0 | 0 | 0 | 10 |
Actually, you may not need to explicitly convert predictions output by celltypist.annotate into an AnnData as above. A more useful way is to use the visualisation function celltypist.dotplot, which quantitatively compares the CellTypist prediction result (e.g. predicted_labels here) with the cell types pre-defined in the AnnData (here cell_type).
[23]:
celltypist.dotplot(predictions, use_as_reference = 'cell_type', use_as_prediction = 'predicted_labels')
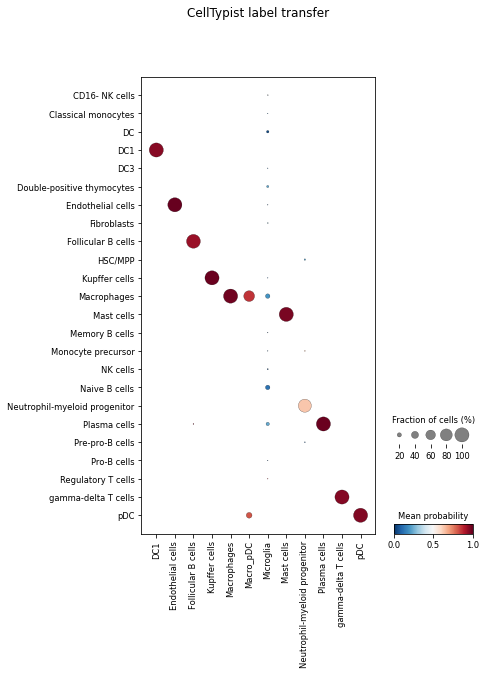
For each pre-defined cell type (each column from the dot plot), this plot shows how it can be ‘decomposed’ into different cell types predicted by CellTypist (rows). You can also change the value of use_as_prediction to majority_voting to compare the majority-voting result with the pre-defined cell types.
[24]:
celltypist.dotplot(predictions, use_as_reference = 'cell_type', use_as_prediction = 'majority_voting')
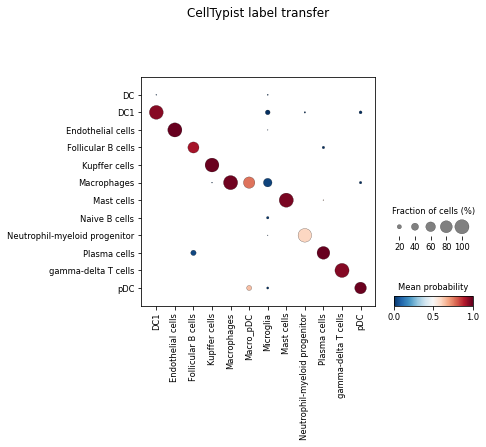
Multi-label classification by utilising a probability threshold
In this section, we show the procedure of transferring multiple cell type labels from built-in models to the query dataset.
All cell types from the CellTypist models are trained in an one-vs-rest fashion, resulting in independent probability estimates that can be compared across cell types. Probabilities are transformed from the decision scores by the sigmoid function, and are kept as is without summing up to one for each query cell. Through this, a probability threshold (default to 0.5, p_thres = 0.5) can be used to determine the cell type(s) assigned to a given cell.
Turn on the multi-label classification by setting the mode = 'prob match' argument.
[25]:
# `p_thres` defaults to 0.5.
predictions = celltypist.annotate(adata_500, model = 'Immune_All_Low.pkl', majority_voting = True, mode = 'prob match', p_thres = 0.5)
🔬 Input data has 500 cells and 18950 genes
🔗 Matching reference genes in the model
🧬 4715 features used for prediction
⚖️ Scaling input data
🖋️ Predicting labels
✅ Prediction done!
👀 Detected a neighborhood graph in the input object, will run over-clustering on the basis of it
⛓️ Over-clustering input data with resolution set to 5
🗳️ Majority voting the predictions
✅ Majority voting done!
[26]:
adata = predictions.to_adata()
[27]:
sc.tl.umap(adata)
[28]:
sc.pl.umap(adata, color = ['cell_type', 'majority_voting'], legend_loc = 'on data')

With the mode of probabilistic match, Microglia is predicted as Unassigned, and Macro_pDC is predicted as Macrophages|pDC (which follows the naming scheme of celltype1|celltyp2)
The probability estimates can be inserted into the adata as well by setting insert_prob = True in the to_adata function. After the insertion, multiple columns will show up in the cell metadata, with each column’s name being a cell type name (no prefix by default) representing probabilities of this cell type distributed across the query cells.
[29]:
adata = predictions.to_adata(insert_prob = True)
adata.obs[['cell_type', 'Plasma cells']]
[29]:
| cell_type | Plasma cells | |
|---|---|---|
| cell1 | Plasma cells | 9.963127e-01 |
| cell2 | Plasma cells | 9.994784e-01 |
| cell3 | Plasma cells | 9.999570e-01 |
| cell4 | Plasma cells | 9.960702e-01 |
| cell5 | Plasma cells | 9.988881e-01 |
| ... | ... | ... |
| cell496 | Macro_pDC | 4.877905e-06 |
| cell497 | Macro_pDC | 5.033182e-07 |
| cell498 | Macro_pDC | 3.668227e-06 |
| cell499 | Macro_pDC | 5.105727e-06 |
| cell500 | Macro_pDC | 6.045739e-06 |
500 rows × 2 columns
[30]:
sc.pl.umap(adata, color = ['cell_type', 'Macrophages', 'pDC'], vmin = 0, vmax = 1, legend_loc = 'on data')

Examination of probability distributions of Macrophages and pDC shows the co-existence of their signatures in the doublet cluster Macro_pDC, as well as noticeable Macrophages scores in the Microglia cluster. Thus even CellTypist assigns the Microglia as Unassigned, the probability scores still indicate their possible transcriptomic similarity with Macrophages.
Multi-label classification using a custom model
In this section, we show the procedure of generating a custom model and transferring multiple labels from the model to the query data.
Download a dataset of 2,000 immune cells as the training set.
[31]:
adata_2000 = sc.read('celltypist_demo_folder/demo_2000_cells.h5ad', backup_url = 'https://celltypist.cog.sanger.ac.uk/Notebook_demo_data/demo_2000_cells.h5ad')
Use previously downloaded scRNA-seq dataset of 500 immune cells as a query.
[32]:
adata_500 = sc.read('celltypist_demo_folder/demo_500_cells.h5ad', backup_url = 'https://celltypist.cog.sanger.ac.uk/Notebook_demo_data/demo_500_cells.h5ad')
Derive a custom model by training the data using the celltypist.train function.
[33]:
# The `cell_type` in `adata_2000.obs` will be used as cell type labels for training.
new_model = celltypist.train(adata_2000, labels = 'cell_type', n_jobs = 10, feature_selection = True)
🍳 Preparing data before training
✂️ 2749 non-expressed genes are filtered out
⚖️ Scaling input data
🏋️ Training data using SGD logistic regression
🔎 Selecting features
🧬 2619 features are selected
🏋️ Starting the second round of training
🏋️ Training data using logistic regression
✅ Model training done!
Refer to the function celltypist.train for what each parameter means, and to the usage for details of model training.
This custom model can be manipulated as with other CellTypist built-in models. First, save this model locally.
[34]:
# Save the model.
new_model.write('celltypist_demo_folder/model_from_immune2000.pkl')
You can load this model by models.Model.load.
[35]:
new_model = models.Model.load('celltypist_demo_folder/model_from_immune2000.pkl')
Next, we use this model to predict the query dataset of 500 immune cells.
[36]:
# Not run; predict the identity of each input cell with the new model.
#predictions = celltypist.annotate(adata_500, model = new_model, majority_voting = True, mode = 'prob match', p_thres = 0.5)
# Alternatively, just specify the model path (recommended as this ensures the model is intact every time it is loaded).
predictions = celltypist.annotate(adata_500, model = 'celltypist_demo_folder/model_from_immune2000.pkl', majority_voting = True, mode = 'prob match', p_thres = 0.5)
🔬 Input data has 500 cells and 18950 genes
🔗 Matching reference genes in the model
🧬 2619 features used for prediction
⚖️ Scaling input data
🖋️ Predicting labels
✅ Prediction done!
👀 Can not detect a neighborhood graph, will construct one before the over-clustering
⛓️ Over-clustering input data with resolution set to 5
🗳️ Majority voting the predictions
✅ Majority voting done!
[37]:
adata = predictions.to_adata(insert_prob = True)
[38]:
sc.tl.umap(adata)
[39]:
sc.pl.umap(adata, color = ['cell_type', 'majority_voting'], legend_loc = 'on data')

Again, Microglia is predicted as Unassigned, and Macro_pDC is predicted as Macrophages|pDC.
[40]:
adata.obs.loc[adata.obs.cell_type == 'Microglia', new_model.cell_types].plot(kind = 'box', rot = 90, figsize = [8, 4], title = 'Microglia')
adata.obs.loc[adata.obs.cell_type == 'Macro_pDC', new_model.cell_types].plot(kind = 'box', rot = 90, figsize = [8, 4], title = 'Macro_pDC')
[40]:
<AxesSubplot:title={'center':'Macro_pDC'}>
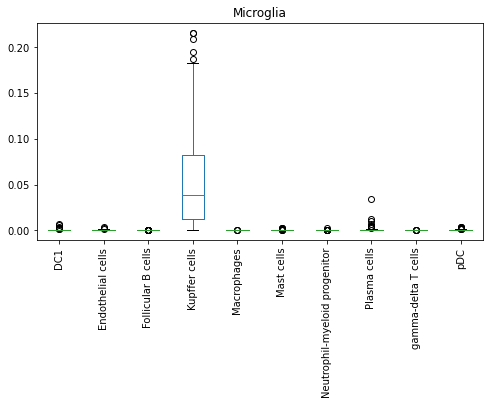
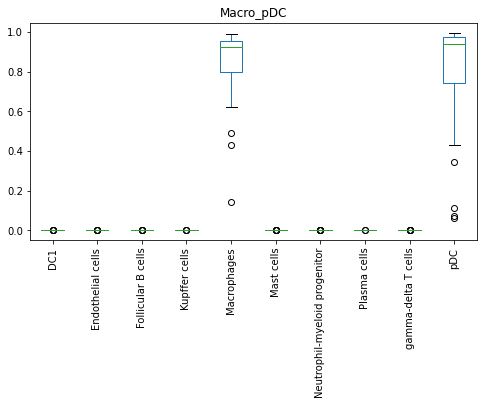
Based on this model, Microglia, though designated as Unassigned by CellTypist, holds relatively higher probability scores with Kupffer cells, signifying a possible tissue-resident macrophage type. Macro_pDC, on the other hand, holds relatively higher probability scores with both Macrophages and pDC.
Examine expression of cell type-driving genes
Each model can be examined in terms of the driving genes for each cell type. Note these genes are only dependent on the model, say, the training dataset.
[41]:
# Any model can be inspected.
# Here we load the previously saved model trained from 2,000 immune cells.
model = models.Model.load(model = 'celltypist_demo_folder/model_from_immune2000.pkl')
[42]:
model.cell_types
[42]:
array(['DC1', 'Endothelial cells', 'Follicular B cells', 'Kupffer cells',
'Macrophages', 'Mast cells', 'Neutrophil-myeloid progenitor',
'Plasma cells', 'gamma-delta T cells', 'pDC'], dtype=object)
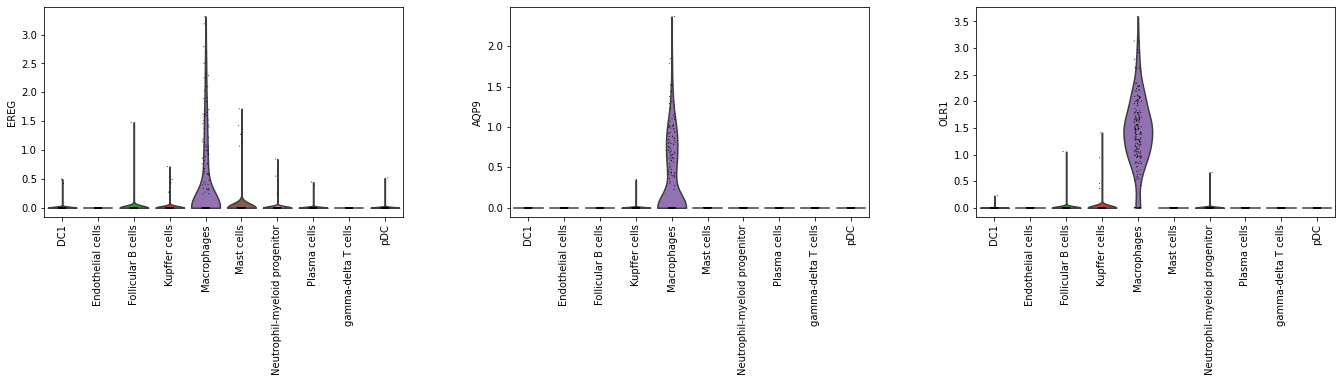
Extract the top three driving genes of Macrophages using the extract_top_markers method.
[43]:
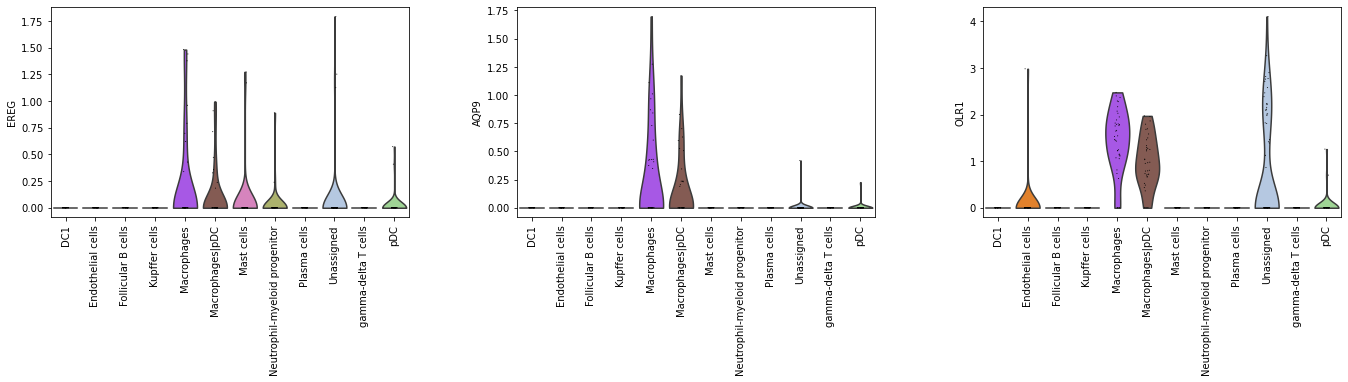
top_3_genes = model.extract_top_markers("Macrophages", 3)
top_3_genes
[43]:
array(['EREG', 'AQP9', 'OLR1'], dtype=object)
[44]:
# Check expression of the three genes in the training set.
sc.pl.violin(adata_2000, top_3_genes, groupby = 'cell_type', rotation = 90)
[45]:
# Check expression of the three genes in the query set.
# Here we use `majority_voting` from CellTypist as the cell type labels for this dataset.
sc.pl.violin(adata_500, top_3_genes, groupby = 'majority_voting', rotation = 90)